
Medical devices and tissue engineered products can be made of same biomaterial, but are they regulated in the same way?
What do wound care products, fillers, and contact lenses have in common? They are all healthcare products that can be made from the same biomaterial. What are biomaterials and is their path to a commercial product always the same?
There are many types of products used in healthcare and they have many purposes. These products are heavily regulated, and their safety and effectiveness have to be monitored throughout their product life cycle. The requirements the product must meet to enter the market are mainly determined by the product’s intended purpose and function. This article provides an overview on key issues when determining which regulation needs to be followed when developing different products used in healthcare.
Biomaterials
Biomaterials are substances or materials that are engineered and designed to interact with biological systems, such as living cells, tissues, and organs. These materials are used in various medical and healthcare applications, such as implants, drug delivery systems, tissue engineering scaffolds, and regenerative medicine. Sometimes, the word ”medical” is added in front of the word “biomaterial” to emphasize the use of biomaterials in medical applications.
There are various types of biomaterials, and new ones are constantly being developed. Some commonly used biomaterial groups in medical applications include metals, ceramics, polymers, and composites. Biomaterials can be derived from natural sources, synthetic materials, or a combination of both. They can also be classified based on key properties such as biocompatibility, bioresorbability, chemical or mechanical characteristics, and their ability to perform specific functions within the body. The properties of biomaterials are modified using different processing methods to create products suitable for specific applications.
When a product made of biomaterial is wanted on the market, it almost always requires marketing approval. To obtain approval, the product must meet the requirements set by the regulations of the specific market area. The requirements are usually determined based on the intended purpose of the product. This means that depending on the intended purpose of the product, products made of the same biomaterial may have very different requirements and marketing approval processes.
Medical Device, Medicinal Product, ATMP, or a combination?
The starting point to knowing which regulation to follow is to know the product’s intended purpose and its operating principle.. Since many requirements apply to products throughout their entire life cycle, it is important to try to know your own product as well as possible from the very beginning of the development work.
One important term when making a product description is intended purpose. It means the use for which a device is intended, according to the data supplied by the manufacturer on the label, in the instructions for use or in promotional or sales materials or statements and as specified by the manufacturer in the clinical evaluation. Sometimes the term intended purpose is confused with the term indications for use, which means the conditions or reasons for using the device.
Another important piece of information is the way in which the intended purpose is achieved. If the intended use is achieved in a pharmacological, metabolic, or immunological way, the product is most likely a medicinal product. In that case, Directive 2001/83/EC is followed in the EU. If it is achieved, for example, by physical, simple chemical, mechanical or digital means, the product is a medical device and then Regulation (EC) No 2017/745 or No 2017/746 has to be complied with. Tissue engineered products, i.e., those that contain cells or tissues that have been modified in such a way that they can be used to repair, regenerate, or replace human tissue, belong to advanced therapy medicinal products (ATMP) in the EU. In these cases, Regulation (EC) No 1394/2007 is followed.
But which regulation is followed when the product has two modes of actions, one of which is achieved mechanically and the other pharmacologically? In that case, the manufacturer defines which is the primary and which is the secondary mode of action. The primary mode of action determines the regulation to be followed.
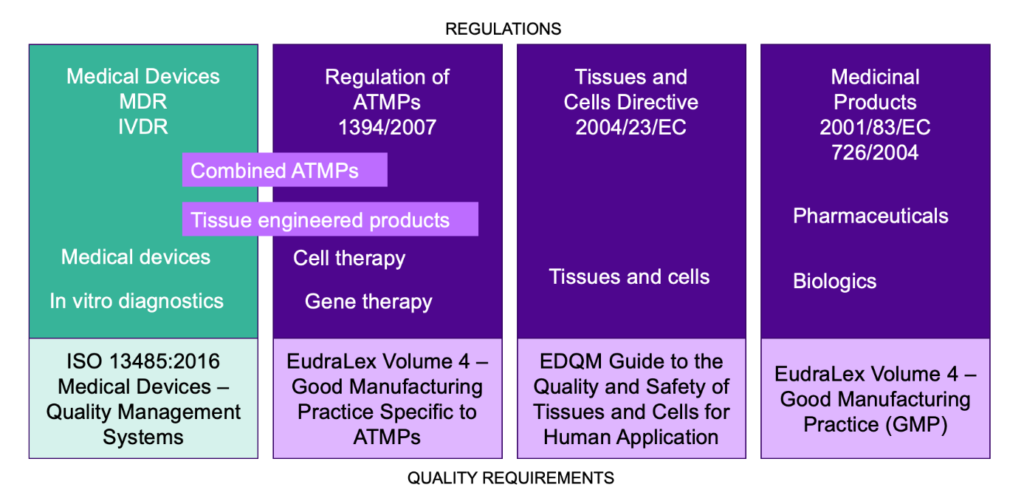
Different Regulatory Processes
When looking at the regulations regarding medical devices and medicines, you may notice that the marketing approval processes they describe are different, as are the authorities. Below, some key differences in these processes for medical devices and ATMPs in the EU are listed.
1. Definition and Scope
a. Medical devices: Medical devices are defined as any instrument, apparatus, implant, software, or other similar or related product used for medical purposes, including diagnosis, prevention, monitoring, treatment, or alleviation of disease. They range from simple tools like bandages to complex devices like MRI machines.
b. ATMPs: ATMPs are a special category of medicinal products that include gene therapies, somatic cell therapies, and tissue-engineered products. These products are often innovative and involve the use of cells, genes, or tissues to provide therapeutic effects.
2. Regulatory Classification
a. Medical devices: Medical devices are classified into four risk-based classes: I, IIa, IIb and III. Requirements the product must meet are based on classification. The higher the class, the more requirements.
b. ATMPs: There is no similar classification system with ATMPs as medical devices have. ATMPs are generally considered high-risk products.
3. Regulatory Approvals
a. Medical devices: The regulatory approval process for medical devices involves conformity assessment procedures based on the device’s risk classification. Manufacturers can place a CE mark on a device once it has passed a conformity assessment. Notified bodies are the only recognized third party bodies that can carry out a conformity assessment.
b. ATMPs: The approval process for ATMPs is more stringent. It requires an extensive evaluation of the quality, safety, and efficacy of the product. ATMPs undergo centralized authorization through the European Medicines Agency (EMA). The evaluation involves scientific assessment by the Committee for Advanced Therapies (CAT) and approval by the European Commission.
4. Clinical Evaluation
a. Medical devices: Medical devices require a clinical evaluation to assess their safety and performance. The level of clinical evidence required depends on the device’s risk class. For higher-risk devices, clinical investigations may be necessary.
b. ATMPs: ATMPs also require robust clinical evidence, but the nature of the evidence differs due to the complexity of the products. Clinical trials play a crucial role in demonstrating the safety and efficacy of ATMPs. The trials need to follow specific guidelines and requirements established by regulatory authorities.
5. Post-Market Surveillance
a. Medical devices: Post-market surveillance is an ongoing process for medical devices. Manufacturers must actively monitor the safety and performance of their devices once they are on the market and report any adverse events or incidents. Notified Bodies also play a role in surveillance activities.
b. ATMPs: Similar to medical devices, post-market surveillance is essential for ATMPs. Manufacturers are required to monitor the safety and effectiveness of the products post-authorization and report any adverse events. Regulatory authorities closely monitor the safety and performance of ATMPs through pharmacovigilance systems.
In conclusion, biomaterials are a broad group of different materials that can be used in a wide range of medical and healthcare applications. Products made from biomaterials used in those applications are heavily regulated, and in order to know what regulation is being followed, you need to know the purpose of the product and the way it is achieved. This knowledge will get you far on your journey towards a commercial product. Both medical devices and tissue-engineered products can be made from the same biomaterial, but the intended purpose indicates which regulations to be followed.
Author: Veiranto Minna, Juusela Maiju, Kellomäki Minna
References
Regulation (EU) 2017/745 of the European Parliament and of the Council of 5 April 2017 on medical devices. Available at: https://eur-lex.europa.eu/legal-content/EN/TXT/?uri=CELEX%3A32017R0745
Regulation (EU) 2017/746 of the European Parliament and of the Council of 5 April 2017 on in vitro diagnostic medical devices. Available at: https://eur-lex.europa.eu/eli/reg/2017/746/oj
Regulation (EC) No 1394/2007 of the European Parliament and of the Council of 13 November 2007 on advanced therapy medicinal products. Available at: https://eur-lex.europa.eu/legal-content/EN/ALL/?uri=celex%3A32007R1394
Directive 2001/83/EC of the European Parliament and of the Council of 6 November 2001 on the Community code relating to medicinal products for human use. Availabe at: https://eur-lex.europa.eu/legal-content/EN/TXT/?qid=1410944582971&uri=CELEX:02001L0083-20121116
Advanced therapy medicinal products: Overview. Available at: https://www.ema.europa.eu/en/human-regulatory/overview/advanced-therapy-medicinal-products-overview
Keywords
biomaterial, medical device, advanced therapy medicinal product, intended purpose, MDR, ATMP, regulatory process, EU regulation