From Definitions to Regulations of Medical devices and Materials
Definitions
One of the main problems while working with biomaterials and medical devices is to find proper definitions for related terminologies, and often those definitions may not represent an agreement among stakeholders or have legal basis. Biomaterials and medical devices are separate entities related to each other, meaning that a single biomaterial can act as a medical device, but medical devices can be made from multiple separate or mixtures of natural or synthetic materials.Biomaterials
The National Institutes of Health Consensus Development Conference Statement on the Clinical Applications of Biomaterials. November 1-3, 1982, defines “biomaterials” as: any substance (other than a drug) or combination of substances, synthetic or natural in origin, which can be used for any time, as whole or as a part of a system, which treats, augments, or replaces any tissue, organ, or function of the body [2].Medical Devices
The US Food and Drug Administration (FDA) defines “medical devices” as follows: Per Section 201(h)(1) of the Food, Drug, and Cosmetic Act, a device is: An instrument, apparatus, implement, machine, contrivance, implant, in vitro reagent, or other similar or related article, including a component part, or accessory which is [3]: (A) recognized in the official National Formulary, or the United States Pharmacopoeia, or any supplement to them, (B) intended for use in the diagnosis of disease or other conditions, or in the cure, mitigation, treatment, or prevention of disease, in man or other animals, or (C) intended to affect the structure or any function of the body of man or other animals, and which does not achieve its primary intended purposes through chemical action within or on the body of humans or other animals and is not dependent upon being metabolized for the achievement of its primary intended purposes. The term “device” does not include software functions excluded pursuant to Section 520(o). The differences between the categories of biomaterials and medical devices are regularly indistinct, because devices are often composed of biomaterials. The FDA category of medical devices involves many entities, including implantable devices and diagnostic-focused items, which are usually not evaluated by toxicologic pathologists. Therefore, it is important to introduce key concepts to evaluate the toxicological pathology of implantable biomaterials and medical devices.Safety
Biocompatibility
The widely accepted definition of biocompatibility is “the ability of a device material to perform with an appropriate host response in a specific situation” [4]. This definition implies that the tissue response is locally controlled at a degree that permits the device to function properly, without unnecessary disturbance of the tissue’s function and structure. Systemic effects also need to be considered when determining biocompatibility, although they are difficult to document because they are rarely identified. “Biocompatibility” usually focuses on the host response to the material using endpoints in toxicologic pathology studies for medical devices, like the degree and relevance of the inflammation, tissue repair, fibrosis, and parenchymal damage. The effects of the host on the device materials should be evaluated according to appropriateness (e.g., biodegradation or mechanical properties), as well as potential adversity and effects of the device materials on the body (e.g., fracture, displacement, unexpected degradation particulates of device material in surrounding and distant tissues, or reduced inflammation due to device material surface modification). The potential adversities also need to be considered for appropriateness (e.g., metal-associated delayed type hypersensitivity reaction) Consequently, biocompatibility should not be considered just in the narrow sense of local appropriate host responses adjacent to implanted device materials but in the whole body.Safety, Function, and Efficacy
The evaluation of the safety, performance, and efficacy of biomaterials and medical devices is usually performed by toxicology and pathology. Safety studies for biomaterials and medical devices are often referred to as biocompatibility studies; nevertheless, individual studies generally provide tolerance indications and adverse reactions by the body under the specific conditions of the study, instead of absolute biocompatibility. In vitro, ex vivo, and in vivo local tissue implantation studies are implemented according to ISO 10993-6:2016 to assess cytotoxicity, genotoxicity, sensitivity, degradation, and local inflammatory and repair responses. For systemic effects, the final safety studies evaluate the medical device in bioassays of relevant location and duration according to ISO 10993-11:2017, including chronic studies, frequently using the final finished form, or intended configuration of the device. These studies are often performed in a clinically relevant surgical animal model (e.g., animals of sufficient body size, such as pigs, and appropriate anatomy, such as monkeys, to model the intended use in humans). The previously described safety studies on the final finished form were also used to evaluate and validate the device function and performance, the work as intended in the intended organ or tissue, and the efficacy, that is, the capacity to treat or prevent diseases. Effectiveness is defined as the performance of a device in a heterogeneous population under real-world conditions, and can only be determined clinically. It is primarily monitored through post-market surveillance and adverse event reporting. Before initiating any clinical study, all animal investigations, including functional and efficacy studies, must be fully completed because of the inability for removal, replacement, or fixation of the high-risk or life-sustaining medical devices from human patients. Clinical studies only involve the target patient population rather than beginning with a cohort of healthy human patients, unlike typical studies on the development of biomolecules or small-molecule products.Integration, Degradation, and Fate of Implanted Medical Materials
The integration or elimination of medical devices involves two opposing processes: encapsulation or tissue ingrowth at the device material, which results in stable fixation and tolerance with functional restoration at the site of the body and its integration. On the other hand, elimination through degradation is generally considered as an indicator for successful medical devices. Biomaterials and medical devices are foreign bodies that do not completely integrate into the host body, organs, or tissues. The degradation of biomaterials is always present, regardless of whether they are categorized as degradable or non-degradable, and they are always subjected to biotribologic forces that drive degradation. Biotribology studies the friction, wear, lubrication, and corrosion of interacting device/tissue surfaces that usually occur between moving body components or between the body and medical materials.Biologic Fate of and Adverse Reactions to Medical Devices
Traditionally, the outcomes or fate of implanted biomaterials or medical devices are limited to an all-in or all-or-none, generally to a local response. Nevertheless, the potential regional and systemic effects and range of fates for permanent and biodegradable medical devices within the body are much broader than those traditionally considered and investigated (Figures 1 and 2). There are numerous biological fates of biomaterials in tissues, such as complete or partial elimination, rejection, encapsulation, resolution of inflammation, return to form/function, stable integration with tissue, replacement of non-regenerating tissue by fibrosis (scar), chronic active inflammation with residues of biomaterials, carcinogenesis, and death from device failure. Minimizing or adjusting tissue reactions is the main goal of biomaterials and medical-device implantation. Surface manipulation or coating of biomaterials with and/or without eluting biologics and drugs are commonly used to enhance the attachment or promote the integration of medical devices into bone and tissues, increase bactericidal action, and minimize inflammatory reactions and fibrosis. [5-7]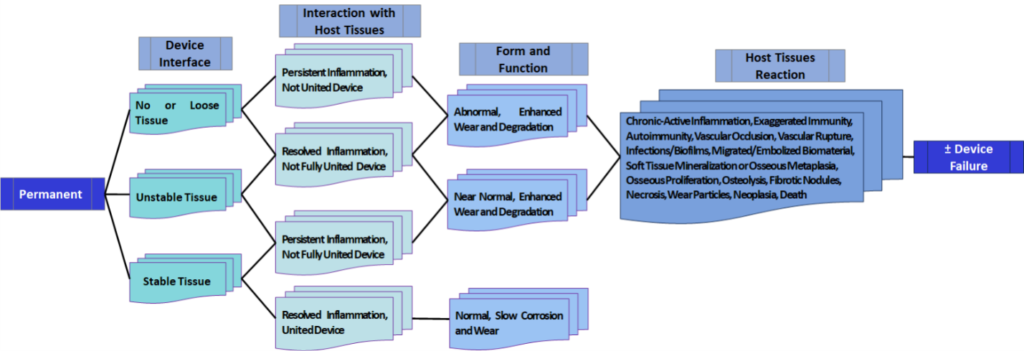
Figure 1. Biologics fates and adverse sequelae to permanent biomaterials and medical devices. Modified from Springer, Integrated Safety and Risk Assessment for Medical Devices and Combination Products by Shayne C. Gad, p. 369. © 2019.
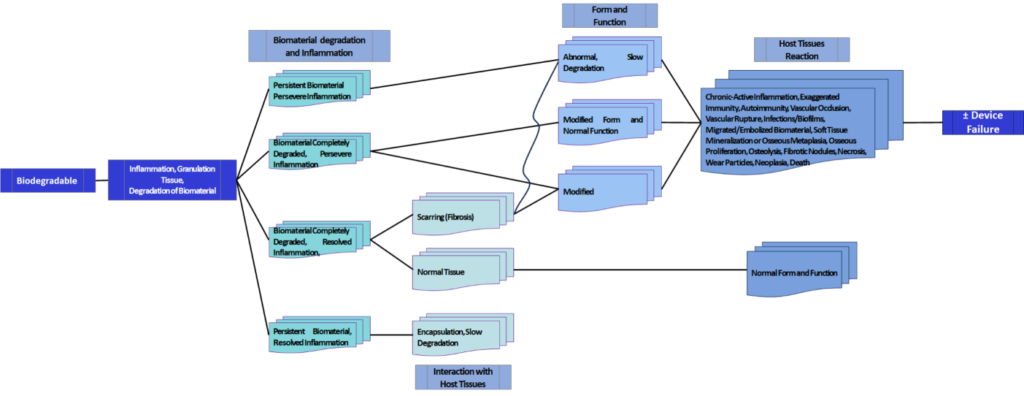
Figure 2. Biologic fates and adverse sequelae to biodegradable biomaterials and medical devices. Modified from Springer, Integrated Safety and Risk Assessment for Medical Devices and Combination Products by Shayne C. Gad, p. 369. © 2019.
Regulations
Regulatory Classification of Devices
Medical device regulation encompasses a multifaceted framework comprising diverse standards, guidelines, and related documents. Due to the dynamic nature of these documents and the evolving nature of laws, it is crucial to continuously review the most up-to-date materials during the design and implementation of medical device studies.
The classification of medical devices is determined based on their risk level. Generally, Class II devices (including IIa and IIb), as well as high-risk devices such as Class III and active implantable medical devices (AIMDs), or Class IV devices in certain countries, are subject to stringent nonclinical and clinical controls and efficacy testing to ensure safety. These devices also necessitate adhere to Good Manufacturing Practices (GMPs) or Quality Management Systems (QMSs).
On the other hand, Class I devices (including Class I basic, I sterile, or I measuring), along with some Class II devices, may undergo self-assessment by the innovator, with minimal in vivo testing for safety and efficacy, provided they demonstrate substantial equivalence to a “predicate device” recently or previously available on the market. Exempt from 510(k) or Premarket Approval (PMA) applications and pre-marketing safety and efficacy approvals are Class I devices. However, Class II devices require GMP and may necessitate unique controls to demonstrate safety and efficacy based on their intended use. For instance, intraocular lens guides are not exempt if used as a folder or injector for soft and foldable intraocular lenses.
In various countries and areas, including the European Union (EU), European Economic Area (EEA), United Kingdom, Canada, Australia, and New Zealand, only nonsterile, non-measuring Class I devices are exempt from regulatory applications before registration and manufacturing. However, post-marketing surveillance may still be required.
While device classification can often be determined by referring to predicate devices, it is essential to consult country-specific information to verify device classification and the appropriate testing approach. Novel biomaterials or devices may necessitate direct engagement with regulatory agencies. Moreover, to ensure product safety, medical device registration might require a country-specific unique device identification (UDI) and a standardized device group descriptor and code, employing the Global Medical Device Nomenclature (GMDN).
The UDI system is adopted by the United States, China, South Korea, Saudi Arabia, and Taiwan, with its introduction currently underway in the EU and the United Kingdom. The GMDN is utilized by the FDA, Canada, EU, Brazil, Australia, New Zealand, Russia, Saudi Arabia, Turkey, and more recently in the United Kingdom as part of its separation from the EU (Brexit). The EU favours the European Medical Device Nomenclature, which is cross-referenced with the GMDN for comprehensive code searching.
Regulatory Standards
The standards for medical devices have experienced independent development compared to those for pharmaceuticals and biologics. Most of the medical device development is directed by international and country-specific testing standards organizations, such as ASTM (American Society for Testing and Materials) and ISO (International Organization for Standardization), rather than the International Council for Harmonisation (ICH) or individual government regulatory agency guidelines.
A recent comprehensive review has examined the current standards and regulatory guidance documents relevant to the development of medical devices. [8] It is important that the standards and regulatory control of medical devices are rapidly evolving. Therefore, it is essential to consult the most up-to-date versions of existing standards and any new standards or regulatory guidelines when designing and reporting studies.
The issuance of these standards and regulatory guidelines plays a crucial role in ensuring the development of safe, effective, and high-quality medical devices. They provide a framework for manufacturers, researchers, and regulatory bodies to establish consistent practices, evaluate device performance, and protect public health. Given the dynamic nature of standards and regulations in this field, it is imperative to stay informed about the latest developments to ensure adherence and compliance in study design and reporting for medical device development.
Note: This article is based on Wancket, L.M., J.C. Schuh, and E. Drevon-Gaillot, Biomedical materials and devices, in Haschek and Rousseaux’s Handbook of Toxicologic Pathology, Volume 2: Safety Assessment Environmental Toxicologic Pathology. 2023, Elsevier
Authors: Juan Uribe-Gomez, Bohara Raghvendra
References
[1] Wancket, L.M., J.C. Schuh, and E. Drevon-Gaillot, Biomedical materials and devices, in Haschek and Rousseaux’s Handbook of Toxicologic Pathology, Volume 2: Safety Assessment Environmental Toxicologic Pathology. 2023, Elsevier. p. 427-466.
[2] Zhang X, W.D., National institutes of health consensus development conference statement on the clinical applications of biomaterials. November 1-3, 1982, in Artif Organs. 1983. p. 260-5.
[3] Food, U. and D. Administration, How to determine if your product is a medical device. US Food and Drug Administration, White Oak, MD, accessed Jun, 2023. 20.
[4] Zhang, X. and D. Williams, Definitions of biomaterials for the twenty-first century. 2019: Elsevier.
[5] Klopfleisch, R. and F. Jung, The pathology of the foreign body reaction against biomaterials. Journal of biomedical materials research Part A, 2017. 105(3): p. 927-940.
[6] Morais, J.M., F. Papadimitrakopoulos, and D.J. Burgess, Biomaterials/tissue interactions: possible solutions to overcome foreign body response. The AAPS journal, 2010. 12: p. 188-196.
[7] Tang, L., P. Thevenot, and W. Hu, Surface chemistry influences implant biocompatibility. Current topics in medicinal chemistry, 2008. 8(4): p. 270-280.
[8] Schuh, J.C. and K.A. Funk, Compilation of international standards and regulatory guidance documents for evaluation of biomaterials, medical devices, and 3-D printed and regenerative medicine products. Toxicologic Pathology, 2019. 47(3): p. 344-357.
Keywords
definitions of biomaterials, definitions of medical devices, the regulatory guidelines, the safety of biomaterials, safety of medical devices, functional assessments of medical devices, organ systems, regulations