
Approval of medical implants according to the European Medical Device Regulation (MDR)
Medical implants for human applications are subject to high legal requirements to ensure safety and performance. As of May 26, 2021, the new European Medical Device Regulation (EU)2017/7458 MDR) [1], [2] also applies to implants. This article outlines the regulatory requirements for medical implants in Europe as well as the connected procedure.
CE-Certification
The CE mark is a prerequisite for launching a new implant in the EU. Before marketing, a manufacturer must first and foremost prove that the implant is safe and efficient and has a positive benefit-risk ratio. A notified body tests and certifies this proof. Only then may the manufacturer affix the CE mark and place the implant on the market. With regards to the requirements under medical device law, the following essential steps can be differentiated on the way to placing a product on the market and the downstream market phase: 1. qualification of the implant as a medical device; 2. risk classification of the implant; 3. manufacturer’s obligations; 4. product requirements; 5. declaration of conformity; 6. CE marking; 7. registration and 8. Monitoring [3].
Qualification of the implant as a medical product
The manufacturer must first determine whether their implant is a medical product as defined in Art. 2 (1) MDR on the basis of its intended purpose. Other characteristics of a medical device are that it is intended for use in humans, its principal action takes place in or on the body, and that this is not achieved by pharmacological or immunological means or metabolically. If the implant in question is a medical device, both the device and the manufacturer are subject to the provisions of the MDR. It should be noted that the scope of the MDR now also includes products without a medical purpose, such as coloured contact lenses or implants and substances for aesthetic purposes. The further text of the MDR does not speak of “implant”, but of “implantable device”, which is defined in Art. 2 (5) MDR as follows: “a device, even if it is intended to be completely or partially absorbed, which is intended to be completely introduced into the human body by a clinical intervention or to replace an epithelial surface or the surface of the eye and to remain there after the intervention. An implantable device also means any device that is intended to be partially introduced into the human body by clinical intervention and to remain there for at least 30 days after the intervention.” [3]
Risk classification of the implant
Manufacturers must classify their medical devices into classes I, IIa, IIb and III, taking into account the intended purpose and the associated risks (Art. 51 MDR). The risk class is the basis for the further steps up to CE marking, in particular for the choice of the conformity assessment procedure and for the scope of the clinical evaluation. Under the previous European regulatory framework, implants were classified according to Annex IX of Directive 93/42/EEC supplemented by Directive 2007/47/EC. In the body periphery, implants were generally assigned to Class IIb and, for the cardiac, central circulatory and nervous system site of application, to Class III [4]. Directives 2003/12/EC and 2005/50/EC specified Class III classification for breast and joint implants. An exception were denture implants, which were assigned to Class IIa. Guidance for the classification of medical devices according to MDR is provided by the MDCG Medical Device Coordination Group Guideline 2021-24 [5]. This also contains examples of risk classes assigned in each case.
If the manufacturer uses software that controls or influences the use of an implant and neither has its own medical purpose nor generates independent information for a medical purpose, MDCG Guideline 2019-11 must be observed for both qualification as a medical device and classification [6]. In Annex I e) of the study under consideration, a web system for monitoring pacemakers or implantable cardioverter defibrillators is given as an example, which falls within the regulatory scope of the MDR. Of particular importance in connection with the classification of software is, above all, implementation regulation 3.3 in Annex VIII MDR. This states that software that controls a product or influences its use is considered to be in the same class as the product, whereas software that is independent of other products is classified on its own [3].
Manufacturer’s obligations
Regardless of the risk class and type of medical products, all manufacturers are subject to the obligations set forth in Art. 10 MDR. Some of these include fulfilment of essential safety and performance requirements; establishment, documentation, application, maintenance of a risk management system; performance of clinical evaluation including clinical follow-up; preparation and continuous updating of technical documentation; performance of a conformity assessment procedure (including product classification) and preparation of an EU declaration of conformity; establishment of a unique device identification (UDI) system and all necessary registrations of devices and the manufacturer, etc.
Quality management
Manufacturers of medical devices must establish, document, apply, maintain, continuously update and improve a quality management system (QMS). The application of the relevant standard EN ISO 13485 allows manufacturers to implement the legal requirements as far as possible as well as other aspects of the QMS in a meaningful way.
Risk Management
Manufacturers of medical devices must establish, document, apply and maintain a risk management system. The EN ISO 14971 standard is the central standard for the risk management of medical devices. It explains how the corresponding process must be set up and maintained in detail.
Clinical evaluation and testing
The clinical evaluation of medical devices aims to assess and demonstrate the clinical safety and performance of the medical device based on clinical data (Art. 61 and Annex XIV Part A, MDR). The type and scope of this evaluation depend on the intended purpose defined by the manufacturer.
Part of the clinical evaluation is also a post-marketing clinical follow-up (Art. 61 (11) and Annex XIV Part B, MDR).
For implants, clinical trials to generate clinical data are, with some exceptions, mandatory (Art. 61 (4), MDR). The exceptions to this regulation are very narrowly defined. Although it is possible to use data from similar products for clinical evaluation, this has been made considerably more difficult with the introduction of the MDR [7]. Art. 61 (5), MDR also states that even in the case of proven similarity of two class III devices or implants, both manufacturers need a contract that explicitly grants full access to the technical file to the manufacturer of the second device on an ongoing basis.
The provisions for clinical investigations of medical devices in the MDR are very comprehensive and are regulated in Articles 62-82. To support the manufacturers, the EU Commission has published an FAQ list on the subject (MDCG 2021-6, [8]). The type of clinical trial that the manufacturer must choose essentially depends on whether the medical device is CE-marked, used within its intended purpose, or whether additional invasive or patient-invasive procedures are used. The MDR basically distinguishes between three types of clinical trials:
- Clinical trials for conformity assessment according to Art. 62,
- Clinical trials for clinical follow-up according to Art. 74(1), and
- Other clinical trials according to Art. 82.
Information on the safety and performance of the product
According to Art. 39 of the MDR, the legislator writes: “Patients who are implanted with a device should be provided with understandable and easily accessible background information that identifies the implanted device, as well as other relevant information about the device, including any necessary warnings about health risks or precautions that may need to be taken, for example, reference to possible incompatibilities with certain diagnostics or with safety scanners.” This is to be achieved through information on the labelling, packaging and in the instructions for use, as well as through the new implant card.
New for manufacturers is the requirement in the MDR included in Article 18 “Implant card and information to be supplied to the patient with an implanted device “. However, the implant card (IC) is not required for “simple” implants such as sutures, staples, dental fillings, etc. The MDCG published the 2019-8 guidelines for the implementation of Article 18 and 2021-11 on product types [9], [10]. According to the 2019-8 guideline, implant cards should be used for patients to be able to identify the respective implant and to have access to information related to the product, e.g., published in the European database EUDAMED. In addition, patients should be able to identify themselves as requiring special treatment in specific situations, e.g., during security checks. Finally, in emergency situations, the ICwill allow clinical staff or first responders to be informed about the special needs and also precautions to be taken when handling patients.
Technical documentation
The technical documentation is a compilation of all relevant documents of a product. It must be kept up to date during the entire product life cycle. The components of the technical documentation include six essential chapters: 1. product description and specification, 2. information to be supplied by the manufacturer, 3. design and manufacturing information, 4. safety and performance requirements, 5. benefit-risk analysis and risk management, and 6. product verification and validation [3].
Responsible person
Each manufacturer must designate a person responsible for regulatory compliance (Art. 15, MDR). This person must be specially trained and is responsible for [11]:
- The conformity of the medical devices with the regulatory requirements,
- the technical documentation and the EU declaration of conformity, and
- the market surveillance.
These responsibilities must be mapped accordingly by the manufacturer in the processes of the QMS.
Product requirements
The product range of implants is very wide. It includes passive and active implants with application in different body regions for a different duration. In addition, active implants may require control software and programming devices. Therefore, only some of the basic safety and performance requirements for implants are explained below as examples, which may not be applicable to every product in individual cases.
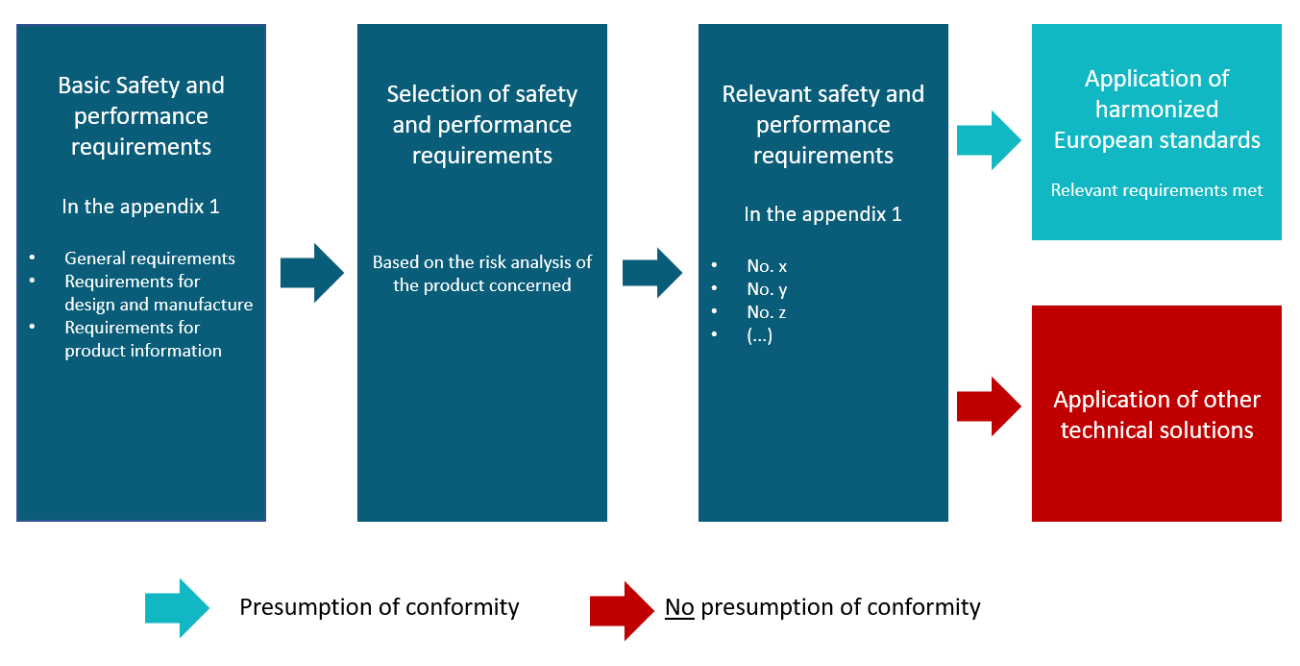
Based on the essential safety and performance requirements (GRUSULA) in Annex I, MDR, the manufacturer makes a selection based on a risk assessment, i.e., an identification of the applicable requirements for the respective product (Figure 1). Compared to the essential requirements in Directive 90/385/EEC, the MDR now also applies many requirements to implants that were previously limited to other medical devices. Other requirements have been retained, such as the specific requirements for active implantable devices in Section 19, MDR.
Custom-made devices, such as those from the dental and dental technology fields, are subject to special requirements. The procedure for custom-made devices is described in Annex XIII, MDR. For example, custom-made devices do not bear a CE mark (Art. 20 (1), MDR) and are subject to a simplified conformity assessment and reduced documentation requirements (Art. 10 (5), MDR).
Information security is also increasingly coming into focus for programmable medical devices or medical products in the form of software, which also applies for some implants. Sections 17.2. and 17.4. in Annex I, MDR require the manufacturer to define risk management including information security as well as minimum requirements for IT security measures. Standards contain detailed descriptions of requirements regarding the technical specifications of medical devices and the relevant processes [14].
The creation of a standard follows a defined process [30]. A distinction is made between different types of standards. In addition to the basic standards (e.g. EN 60601-1) with the general specifications for products, there may also be supplementary standards (e.g. EN 60601-1-1), which describe additional general requirements and tests. Further on, there are product standards (e.g. EN 60601-2-4), also known as vertical standards, which refer to the basic and supplementary standards. They supplement or replace general standards with specific specifications and apply only to certain products in accordance with a European legal act. Procedural or process standards (e.g. EN ISO 13485) describe the requirements for the development, introduction and maintenance of processes and have a cross-product (horizontal) significance.
According to the identified risk class (does not apply to risk class I), the manufacturer can choose among different conformity assessment procedures (Art. 52, Annexes IX-XI, MDR). In the EU type examination, the notified body examines or tests a representative specimen of the product (the so-called type) and the associated technical documentation to determine whether it complies with the legal requirements. Product conformity testing consists of two parts, production quality assurance with an audit by the notified body in relation to production, or the testing of each product to confirm conformity of a particular batch of products. For implants, the full quality assurance system with an audit by the notified body including the review of the technical documentation is likely to be the procedure of choice [3].
Declaration of Conformity
Through the EU declaration of conformity, the manufacturer assumes product responsibility. This is continuously updated and contains specified information according to Annex IV, MDR. In the case of implants that basically belong to risk class IIa or higher, the manufacturer who carries out the conformity assessment procedure, receives a corresponding EU certificate from the notified body and declares conformity [3].
CE conformity mark
The CE conformity mark indicates that the relevant requirements of the MDR have been met. The manufacturer affixes the CE mark with the number of the notified body involved (not for risk class I) on the product, packaging, instructions for use and promotional material. Annex V MDR regulates the appearance of the CE marking [3].
Registration
Before placing the medical device on the market, manufacturers, authorized representatives and importers must register in EUDAMED (Art. 31, MDR). In case of a conformity assessment with the involvement of a notified body, the information (Annex VI Part A Section 1) is transmitted to EUDAMED before the application is submitted to the notified body. The manufacturer must register their devices with the information from Annex VI Part A in EUDAMED (Art. 29, MDR).
As in other medical device markets, the MDR calls for the establishment of a Unique Device Identification (UDI) system (Art. 27, Annex VI, MDR). The primary objectives of the UDI system are to facilitate the traceability of medical devices, to significantly increase the effectiveness of post-market safety-related activities and to enable better monitoring by the competent authorities [3].
Monitoring
MDR medical device surveillance is divided into three measures:
- post-market surveillance by the manufacturer (Arts. 83-86),
- vigilance by the manufacturer (Arts. 87-91), and
- regulatory market surveillance (Arts. 93-100).
The manufacturer must integrate a post-market surveillance system into his QMS (Art. 84 (1), MDR). The related PMS plan and report (Art. 84 and 85, MDR) are components of the technical documentation (3.3.5).
Vigilance of the manufacturer includes reporting of serious incidents and safety corrective actions in the field (Art. 87, MDR). Serious incidents such as death or unforeseen serious deterioration of health must be reported immediately via EUDAMED, no later than ten days after becoming aware of them (Art. 87 (5), MDR). All other serious incidents must also be reported immediately, at the latest within 15 days (Art. 87 (3), MDR).
Although regulatory market surveillance remains a task of the individual member states, it is to be coordinated across the EU. Art. 93 (1), MDR requires competent authorities to inspect the conformity characteristics and performance of medical devices through appropriate reviews of documentation, and through physical checks or laboratory testing of samples. The competent authorities report the results of their monitoring activities (Art. 93 (4), MDR) and file monitoring reports via EUDAMED (Art. 93 (7), MDR) [3].
References (English)
[1] European Parliament and the Council of the European Union, Regulation (EU) 2020/561 amending Regulation (EU) 2017/745 on medical devices as regards the date of application of certain of its provisions. 2020. Available at: https://eur-lex.europa.eu/legal-content/EN/TXT/PDF/?uri=CELEX:32020R0561&from=EN, accessed on 05.10.2022.
[2] European Parliament and Council of the European Union, Regulation (EU) 2017/745 of the European Parliament and of the Council of 5 April 2017 concerning medical devices, amending Directive 2001/83/EC, Regulation (EC) No 178/2002 and Regulation (EC) No 1223/2009 and repealing Council Directives 90/385/EEC and 93/42/EEC. 2017, pp. 1-228. Available at: https://eur-lex.europa.eu/eli/reg/2017/745, accessed on 05.10.2022.
[3] Prinz, T. (2022), “Approval of medical implants under the European Medical Device Regulation (MDR),” RSPONSE Partnership for Innovation in Implant Technology. Available at: https://www.vde.com/resource/blob/2182192/ab8a8c083e21c40e4eecea6d3a86b217/positionspapier-data.pdf, accessed on 05.10.2022.
[4] BVMed – Bundesverband Medizintechnologie e.V., ed., Classification list for medical devices. Berlin, 2014.
[5] Medical Device Coordination Group, MDCG 2021-24 Guidance on classification of medical devices. 2021. Available at: https://health.ec.europa.eu/system/files/2021-10/mdcg_2021-24_en_0.pdf, accessed on 05.10.2022.
[6] Medical Device Coordination Group, MDCG 2019-11 Guidance on Qualification and Classificationof Software in Regulation (EU) 2017/745 – MDR and Regulation (EU) 2017/746 – IVDR. 2019. Available at: https://ec.europa.eu/docsroom/documents/37581, accessed on 05 Oct. 2022.
[7] Medical Device Coordination Group, MDCG 2020-5 Clinical Evaluation – Equivalence. A guide for manufacturers and notified bodies. 2020. Available at: https://health.ec.europa.eu/system/files/2020-09/md_mdcg_2020_5_guidance_clinical_evaluation_equivalence_en_0.pdf, accessed on 05.10.2022.
[8] Medical Device Coordination Group, MDCG 2021-6 Regulation (EU) 2017/745 – Questions & Answers regarding clinical investigation. 2021. Available at: https://health.ec.europa.eu/system/files/2021-04/mdcg_2021-6_en_0.pdf, accessed on 05.10.2022.
[9] Medical Device Coordination Group, MDCG 2019-8 v2 – Guidance document – Implant Card relating to the application of Article 18 Regulation (EU) 2017/745 of the European Parliament and of the Council of 5 April 2017 on medical devices. 2020. Available at: https://ec.europa.eu/health/system/files/2020-09/md_mdcg_2019_8_implant_guidance_card_en_0.pdf, accessed on 05.10.2022.
[10] Medical Device Coordination Group, MDCG 2021-11 Guidance on Implant Card – ‘Device types’.2021. Available at: https://ec.europa.eu/health/sites/default/files/md_sector/docs/md_2021-11_en.pdf, accessed on 05.10.2022.
[11] Medical Device Coordination Group, MDCG 2019-7 Guidance on Article 15 of the Medical Device Regulation (MDR) and in vitro Diagnostic Device Regulation (IVDR) regarding a ‘person responsible for regulatory compliance’ (PRRC). 2019. Available at: https://ec.europa.eu/docsroom/documents/36166, accessed 05 Oct. 2022, accessed on 05.10.2022.
[12] European Commission, ed, “The ‘Blue Guide’ on the implementation of EU products rules 2016.” July 26, 2016. Available at: https://eur-lex.europa.eu/legal-content/EN/TXT/ PDF/?uri=CELEX:52016XC0726(02)&from=BG, accessed on 05 Oct. 2022.
[13] Global Harmonization Task Force, Role of standards in the assessment of medical devices.
- Available at: http://www.imdrf.org/docs/ghtf/final/sg1/procedural-docs/ghtf sg1-n044-2008-standards-in-assessment-of-medical-devices-080305.pdf, accessed on 05.10.2022.
[14] S. Hallscheidt, N. Adomeit, T. Manske, and J. U. Hopf, “1×1 der Normung – Ein praxis-orientierterLeitfaden für KMU.” May 2019. accessed Sep 19, 2019. Available at: https://www.din.de/blob/64110/b5d8d57f3b7e6866233201bf45aad388/1×1-data.pdf, accessed on 05 Oct. 2022.